
September 2023
(Diana Roopchand)
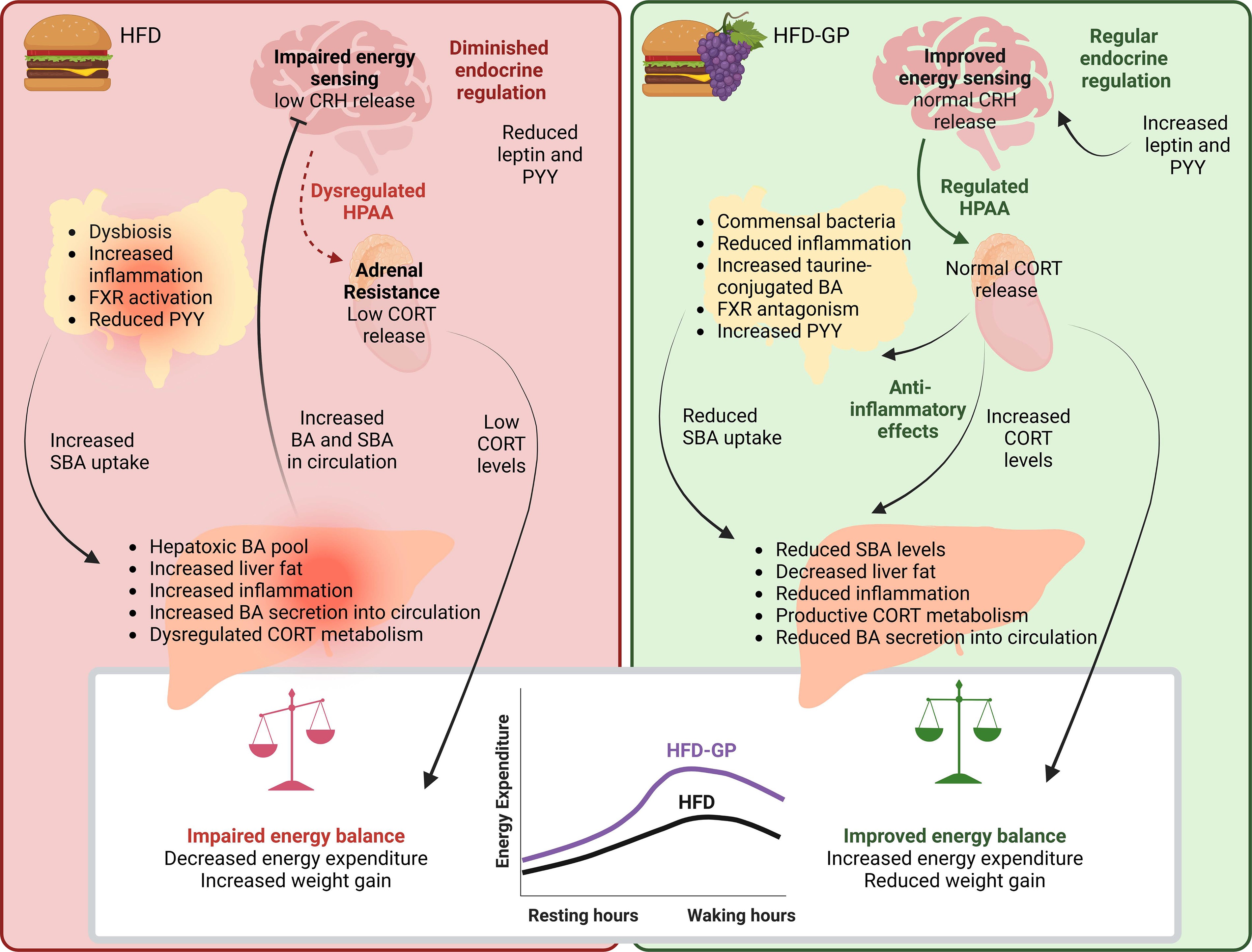
Figure 8. Potential mechanisms for the effects of GPs on HPAA control of energy balance, involving the gut bacteria, BA, and endocrine hormones.
Esther Mezhibovsky, Kevin M Tveter, Jose A Villa-Rodriguez, Karen Bacalia, Dushyant Kshatriya, Nikhil Desai, Alrick Cabales, Yue Wu, Ke Sui, Rocio M Duran, Nicholas T Bello, Diana E Roopchand, Grape Polyphenols May Prevent High-Fat Diet–Induced Dampening of the Hypothalamic–Pituitary–Adrenal Axis in Male Mice, Journal of the Endocrine Society, Volume 7, Issue 9, September 2023, bvad095, https://doi.org/10.1210/jendso/bvad095
Abstract
Context:
Chronic high-fat diet (HFD) consumption causes obesity associated with retention of bile acids (BAs) that suppress important regulatory axes, such as the hypothalamic–pituitary–adrenal axis (HPAA). HFD impairs nutrient sensing and energy balance due to a dampening of the HPAA and reduced production and peripheral metabolism of corticosterone (CORT).
Objective:
We assessed whether proanthocyanidin-rich grape polyphenol (GP) extract can prevent HFD-induced energy imbalance and HPAA dysregulation.
Methods:
Male C57BL6/J mice were fed HFD or HFD supplemented with 0.5% w/w GPs (HFD-GP) for 17 weeks.
Results:
GP supplementation reduced body weight gain and liver fat while increasing circadian rhythms of energy expenditure and HPAA-regulating hormones, CORT, leptin, and PYY. GP-induced improvements were accompanied by reduced mRNA levels of Il6, Il1b, and Tnfa in ileal or hepatic tissues and lower cecal abundance of Firmicutes, including known BA metabolizers. GP-supplemented mice had lower concentrations of circulating BAs, including hydrophobic and HPAA-inhibiting BAs, but higher cecal levels of taurine-conjugated BAs antagonistic to farnesoid X receptor (FXR). Compared with HFD-fed mice, GP-supplemented mice had increased mRNA levels of hepatic Cyp7a1 and Cyp27a1, suggesting reduced FXR activation and more BA synthesis. GP-supplemented mice also had reduced hepatic Abcc3 and ileal Ibabp and Ostβ, indicative of less BA transfer into enterocytes and circulation. Relative to HFD-fed mice, CORT and BA metabolizing enzymes (Akr1d1 and Srd5a1) were increased, and Hsd11b1 was decreased in GP supplemented mice.
Conclusion:
GPs may attenuate HFD-induced weight gain by improving hormonal control of the HPAA and inducing a BA profile with less cytotoxicity and HPAA inhibition, but greater FXR antagonism.
September 2023
(Liping Zhao & Guojun Wu)
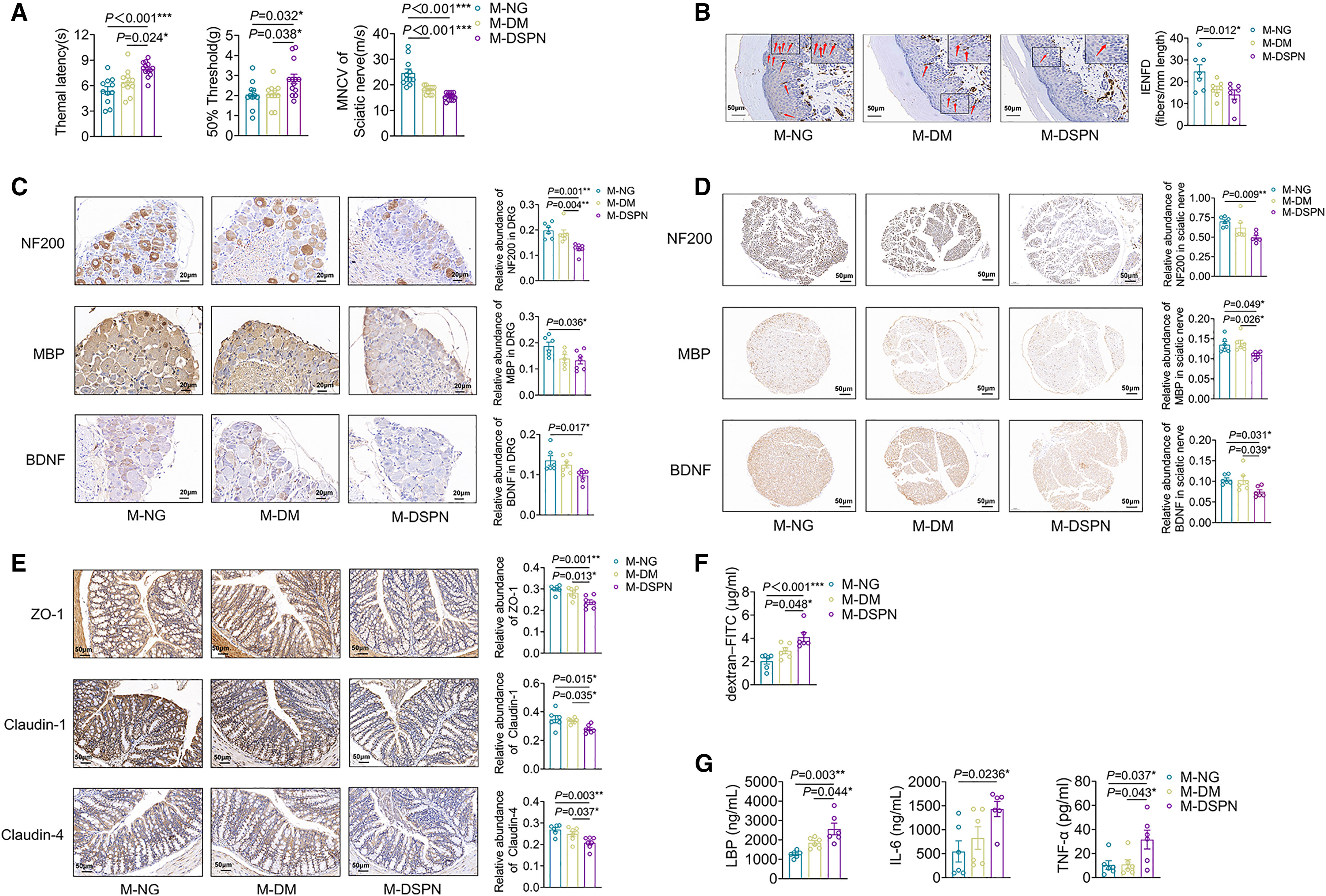
Figure 1 Gut microbiota from patients with DSPN induced more severe peripheral neuropathy than that from the subjects with normal glucose levels or patients with DM but not DSPN in db/db mice
(A) Mechanical sensitivity, thermal sensitivity, and motor nerve conduction velocity (MNCV) of sciatic nerve (M-NG and M-DM groups, n = 11; M-DSPN group, n = 13).
(B–E) Immunohistochemical staining and integrated optical density (IOD) analysis in the three groups.
(B) Intraepidermal nerve fiber density (IENFD, marked by PGP9.5) of the posterior plantar skin (n = 7 per group). Scale bars, 50 μm.
(C and D) Neurofilament 200 (NF200), myelin basic protein (MBP), and brain-derived neurotrophic factor (BDNF) in (C) dorsal root ganglion (for NF200, BDNF, M-NG group, n = 6; M-DM and M-DSPN groups, n = 7; for MBP, M-NG group, n = 6; M-DM group, n = 5; M-DSPN group, n = 7) (scale bars, 20 μm) and (D) sciatic nerve (n = 6 per group; scale bars, 50 μm).
(E) Tight junction proteins ZO-1, Claudin-1, and Claudin-4 in colon (M-NG group, n = 6; M-DM and M-DSPN groups, n = 7). Scale bars, 50 μm.
(F) Plasma level of dextran-FITC (n = 6 per group).
(G) Plasma levels of LBP, IL-6, and TNF-α (n = 6 per group).
M-NG, M-DM, and M-DSPN indicate db/db mice received fecal microbiota from the subjects with normal glucose level, patients with DM but not DSPN, and with DSPN, respectively. Data are presented as the mean ± SEM. The comparison among the three groups was tested by one-way ANOVA, and Tukey’s post hoc test (two-tailed) was used for inter-group comparisons. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Yang, J., Yang, X., Wu, G., Huang, F., Shi, X., Wei, W., Zhang, Y., Zhang, H., Cheng, L., Yu, L., Shang, J., Lv, Y., Wang, X., Zhai, R., Li, P., Cui, B., Fang, Y., Deng, X., Tang, S., Wang, L., … Yuan, H. (2023). Gut microbiota modulate distal symmetric polyneuropathy in patients with diabetes. Cell metabolism, 35(9), 1548–1562.e7. https://doi.org/10.1016/j.cmet.2023.06.010
Abstract
The pathogenic mechanisms underlying distal symmetric polyneuropathy (DSPN), a common neuropathy in patients with diabetes mellitus (DM), are not fully understood. Here, we discover that the gut microbiota from patients with DSPN can induce a phenotype exhibiting more severe peripheral neuropathy in db/db mice. In a randomized, double-blind, and placebo-controlled trial (ChiCTR1800017257), compared to 10 patients who received placebo, DSPN was significantly alleviated in the 22 patients who received fecal microbiota transplants from healthy donors, independent of glycemic control. The gut bacterial genomes that correlated with the Toronto Clinical Scoring System (TCSS) score were organized in two competing guilds. Increased guild 1, which had higher capacity in butyrate production, and decreased guild 2, which harbored more genes in synthetic pathway of endotoxin, were associated with improved gut barrier integrity and decreased proinflammatory cytokine levels. Moreover, matched enterotype between transplants and recipients showed better therapeutic efficacy with more enriched guild 1 and suppressed guild 2. Thus, changes in these two competing guilds may play a causative role in DSPN and have the potential for therapeutic targeting.
August 2023
(Sara Campbell)
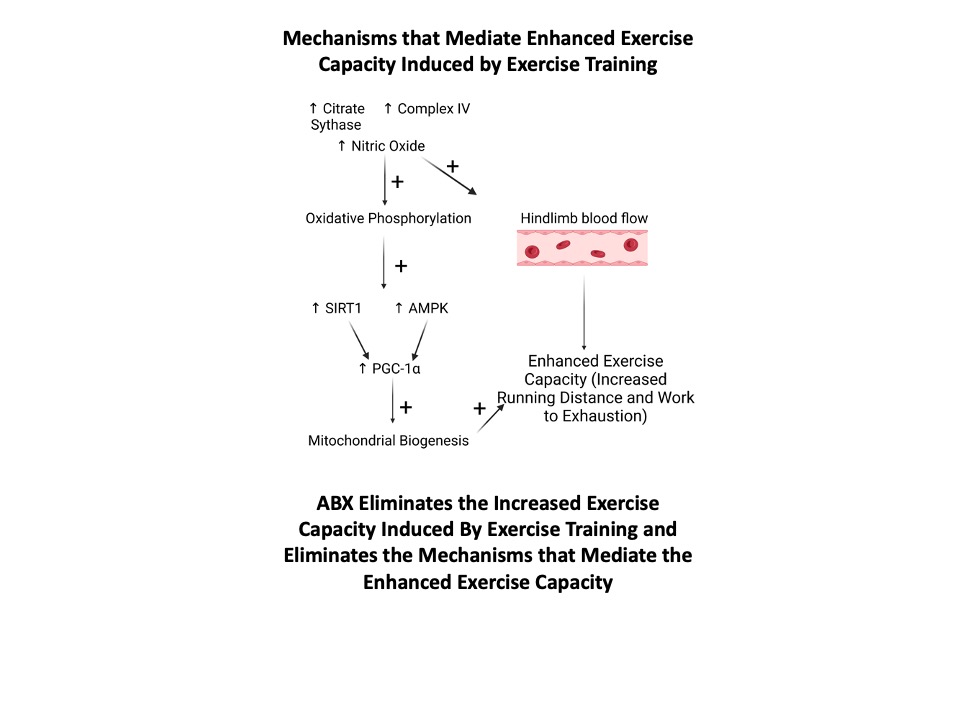
Figure 7 Mechanisms mediating enhanced exercise capacity induced by exercise training. Exercise capacity is increased due to increased hindlimb blood flow mediated by increases nitric oxide and increased mitochondrial oxidative phosphorylation (citrate synthase, complex IV) providing the stimulus to increased mitochondrial biogenesis (AMPK, SIRT1, and PGC-1α). ABX in drinking water abolished the increases in the markers of mitochondrial oxidative function (citrate synthase, complex IV, and nitric oxide) and the increases in mitochondrial biogenesis (AMPK, SIRT1, and PCG-1α). Thus, the elimination of the gut microbiota, using ABX, eliminated the increased running distance and work to exhaustion induced by exercise training and the mechanisms mediating the improved exercise capacity induced by exercise training.
Dowden, R.A, Wisniewski, P.J., Longoria, C.R., Zhang, J. Cavallo, M., Guers, J.J., Oydanich, M., Vater, D.E., Vatner, S.F., Campbell, S.C. (2023). Microbiota Mediate Enhanced Exercise Capacity Induced by Exercise Training. Med Sci Sport Exer. 55(8):1392-1400. (IF – 4.459).
https://journals.lww.com/acsm-msse/pages/default.aspx.
Abstract
The gut microbiota is critical to host metabolism and is influenced by many factors, including host genotype, diet, and exercise training.
Purpose
We investigated the effects of gut microbes, and the mechanisms mediating the enhanced exercise performance induced by exercise training, i.e., skeletal muscle blood flow, and mitochondrial biogenesis and oxidative function in male mice.
Methods
All mice received a graded exercise test before (PRE) and after exercise training via forced treadmill running at 60% to 70% of maximal running capacity 5 d·wk−1 for 5 wk (POST). To examine the role of the gut microbes, the graded exercise was repeated after 7 d of access to antibiotic (ABX)-treated water, used to eliminate gut microbes. Peripheral blood flow, mitochondrial oxidative capacity, and markers of mitochondrial biogenesis were collected at each time point.
Results
Exercise training led to increases of 60% ± 13% in maximal running distance and 63% ± 11% work to exhaustion (P < 0.001). These increases were abolished after ABX (P < 0.001). Exercise training increased hindlimb blood flow and markers of mitochondrial biogenesis and oxidative function, including AMP-activated protein kinase, sirtuin-1, PGC-1α citrate synthase, complex IV, and nitric oxide, all of which were also abolished by ABX treatment.
Conclusions
Our results support the concept that gut microbiota mediate enhanced exercise capacity after exercise training and the mechanisms responsible, i.e., hindlimb blood flow, mitochondrial biogenesis, and metabolic profile. Finally, results of this study emphasize the need to fully examine the impact of prescribing ABX to athletes during their training regimens and how this may affect their performance.
June 2023
(Diana Roopchand)
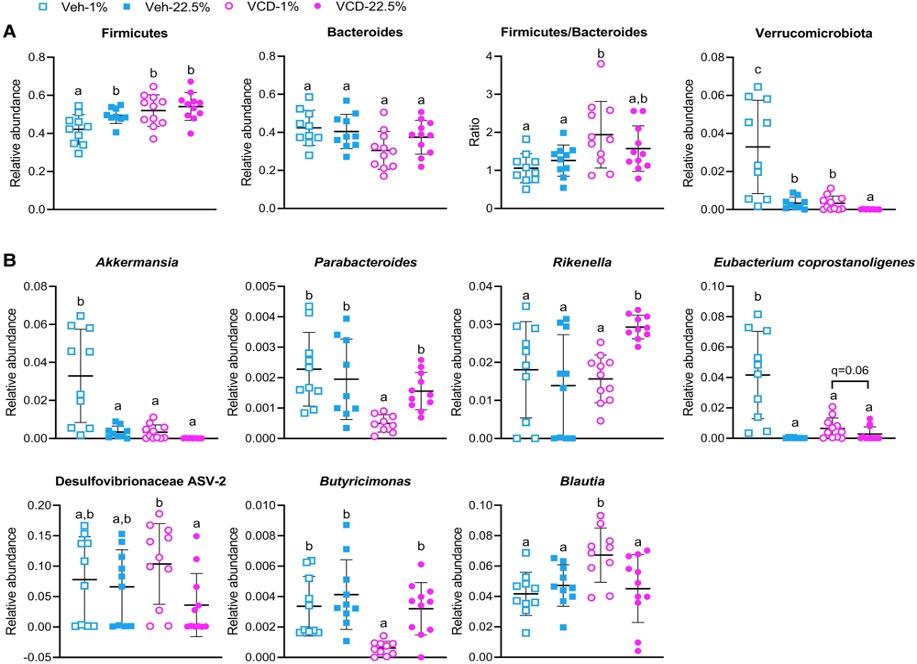
Figure 10. Gut microbial community response to treatment and dietary fatty acids. Relative abundance of amplicon sequence variants (ASVs) identified in fecal gut microbiota samples collected after 23 to 25 weeks of dietary intervention (n = 10 for Veh-treated groups and n = 11 for VCD-treated groups) classified at the (A) phyla and (B) genera level. Different letters (a, b, c) indicate significant difference as determined by the Kruskal–Wallis test followed by 2-stage step-up method of Benjamini, Krieger, and Yekutieli with false discovery rate adjusted P value, q < .05.
Ke Sui, Ali Yasrebi, Natasha Malonza, Zehra H Jaffri, Samuel E Fisher, Isaac Seelenfreund, Brandon D McGuire, Savannah A Martinez, Avery T MacDonell, Kevin M Tveter, Candace R Longoria, Sue A Shapses, Sara C Campbell, Diana E Roopchand, Troy A Roepke, Saturated Fatty Acids and Omega-3 Polyunsaturated Fatty Acids Improve Metabolic Parameters in Ovariectomized Female Mice, Endocrinology, Volume 164, Issue 6, June 2023, bqad059, https://doi.org/10.1210/endocr/bqad059
Abstract
In menopausal and postmenopausal women, the risk for obesity, cardiovascular disease, osteoporosis, and gut dysbiosis are elevated by the depletion of 17β-estradiol. A diet that is high in omega-6 polyunsaturated fatty acids (PUFAs), particularly linoleic acid (LA), and low in saturated fatty acids (SFAs) found in coconut oil and omega-3 PUFAs may worsen symptoms of estrogen deficiency. To investigate this hypothesis, ovariectomized C57BL/6J and transgenic fat-1 mice, which lower endogenous omega-6 polyunsaturated fatty acids, were treated with either a vehicle or estradiol benzoate (EB) and fed a high-fat diet with a high or low PUFA:SFA ratio for ~15 weeks. EB treatment reversed obesity, glucose intolerance, and bone loss in ovariectomized mice. fat-1 mice fed a 1% LA diet experienced reduced weight gain and adiposity, while those fed a 22.5% LA diet exhibited increased energy expenditure and activity in EB-treated ovariectomized mice. Coconut oil SFAs and omega-3 FAs helped protect against glucose intolerance without EB treatment. Improved insulin sensitivity was observed in wild-type and fat-1 mice fed 1% LA diet with EB treatment, while fat-1 mice fed 22.5% LA diet was protected against insulin resistance without EB treatment. The production of short-chain fatty acids by gut microbial microbiota was linked to omega-3 FAs production and improved energy homeostasis. These findings suggest that a balanced dietary fatty acid profile containing SFAs and a lower ratio of omega-6:omega-3 FAs is more effective in alleviating metabolic disorders during E2 deficiency.
June 2023
(Maria Gloria Dominguez-Bello)
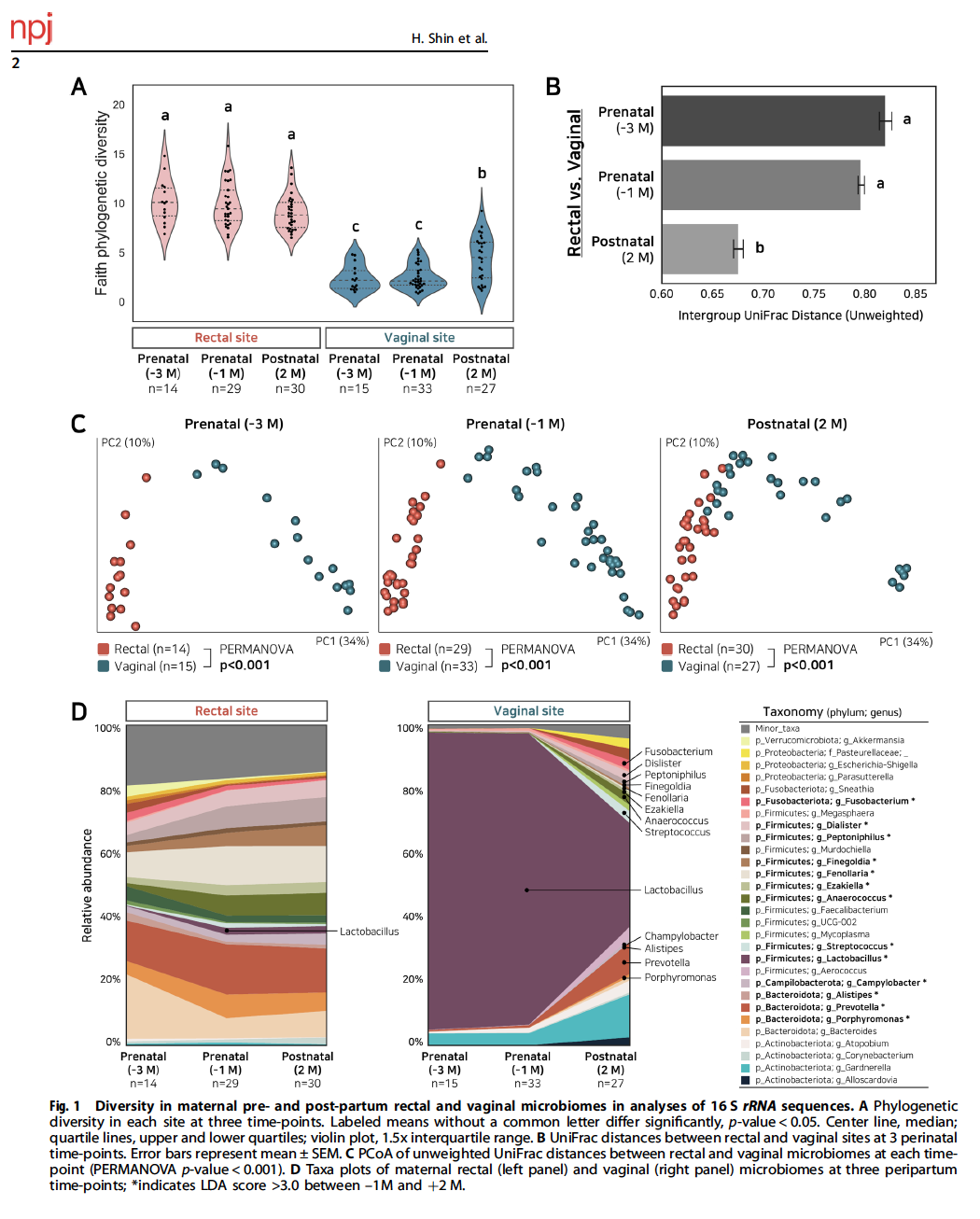
Shin H, Martinez II K, Henderson N, Jay M, Schweizer W, Bogaert D, Park G, Bokulich N, Blaser M., Dominguez Bello MG. Partial convergence of the human vaginal and rectal maternal microbiota in late gestation and the early post-partum" NPJBiofilms Microbiomes, 2023 9:37; https://doi.org/10.1038/s41522-023-00404-5
Abstract:
The human vaginal and fecal microbiota change during pregnancy. Because of the proximity of these perineal sites and theevolutionarily conserved maternal-to-neonatal transmission of the microbiota, we hypothesized that the microbiota of these twosites (rectal and vaginal) converge during the last gestational trimester as part of the preparation for parturition. To test thishypothesis, we analyzed 16S rRNA sequences from vaginal introitus and rectal samples in 41 women at gestational ages 6 and 8months, and at 2 months post-partum. The results show that the human vaginal and rectal bacterial microbiota converged duringthe last gestational trimester and into the 2nd month after birth, with a significant decrease in Lactobacillus species in both sites, asalpha diversity progressively increased in the vagina and decreased in the rectum. The microbiota convergence of the maternalvaginal-anal sites perinatally might hold significance for the inter-generational transmission of the maternal microbiota.npj Biofilms and Microbiomes (2023) 9:37 ; https://doi.org/10.1038/s41522-023-00404-5
April 2023
(Maria Gloria Dominguez-Bello)
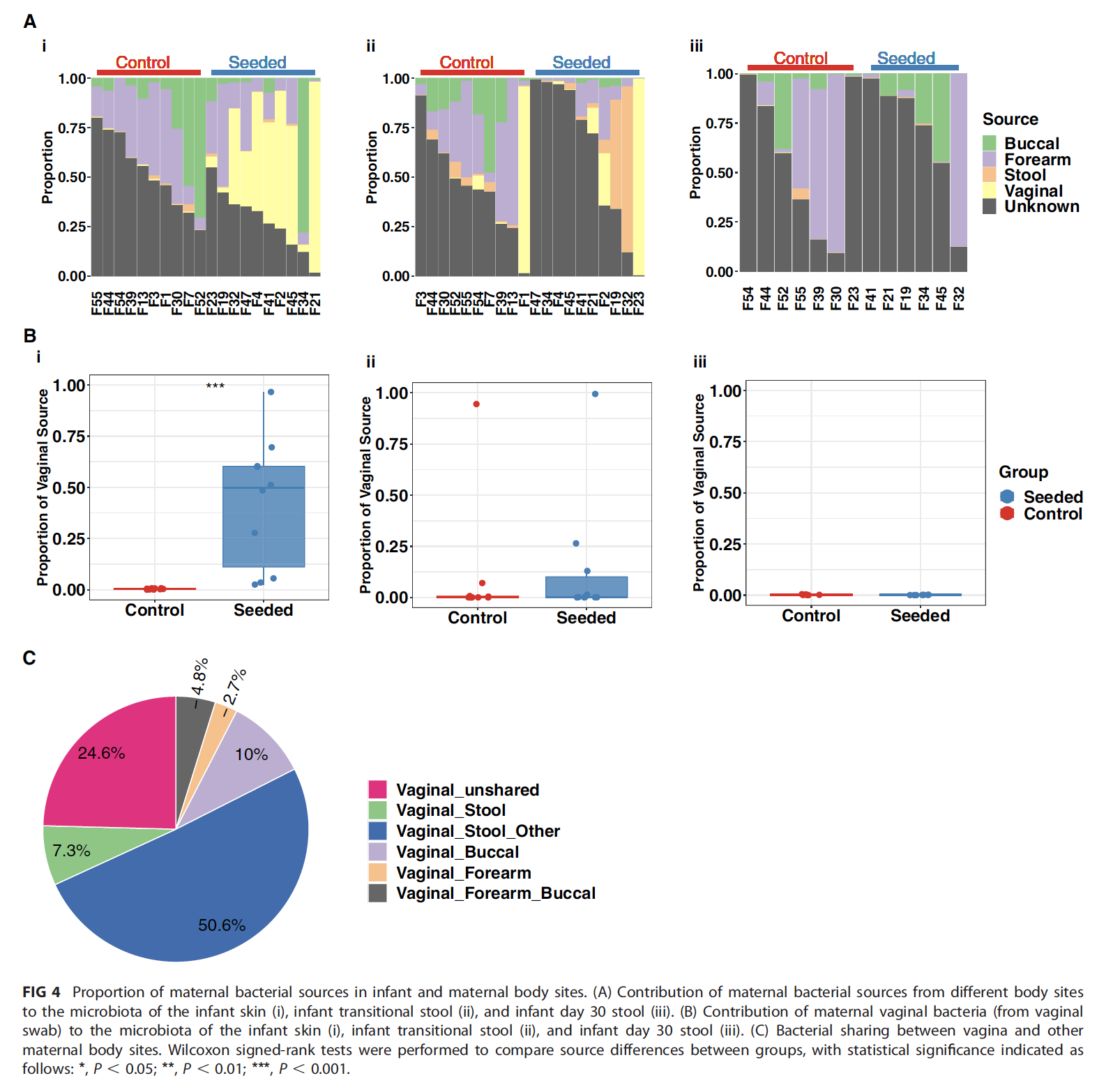
Mueller NT, Differding MK, Sun H, Wang J, Levy S, Deopujari V, Appel LJ, Blaser MJ, Kundu T, Shah AA, Dominguez Bello MG, Hourigan SK. Maternal Bacterial Engraftment in Multiple Body Sites of Cesarean Section Born Neonates after Vaginal Seeding-a Randomized Controlled Trial. mBio, 2023 Apr 19:e0049123. doi: 10.1128/mbio.00491-23. Epub ahead of print. PMID: 37074174.
Abstract:
Children delivered by elective, prelabor Cesarean section (C-section) arenot exposed to the birth canal microbiota and, in relation to vaginally delivered children,show altered microbiota development. Perturbed microbial colonization during criticalearly-life windows of development alters metabolic and immune programming and isassociated with an increased risk of immune and metabolic diseases. In nonrandomizedstudies, vaginal seeding of C-section-born neonates partially restores their microbiota colonizationto that of their vaginally delivered counterparts, but without randomization, confoundingfactors cannot be excluded. In a double-blind, randomized, placebo-controlledtrial, we determined the effect of vaginal seeding versus placebo seeding (control arm)on the skin and stool microbiota of elective, prelabor C-section-born neonates (n = 20)at 1 day and 1 month after birth. We also examined whether there were between-armdifferences in engraftment of maternal microbes in the neonatal microbiota. In relationto the control arm, vaginal seeding increased mother-to-neonate microbiota transmissionand caused compositional changes and a reduction in alpha diversity (Shannon Index) ofthe skin and stool microbiota. The neonatal skin and stool microbiota alpha diversitywhen maternal vaginal microbiota is provided is intriguing and highlights theneed of larger randomized studies to determine the ecological mechanisms andeffects of vaginal seeding on clinical outcomes.
March 2023
(Diana Roopchand)
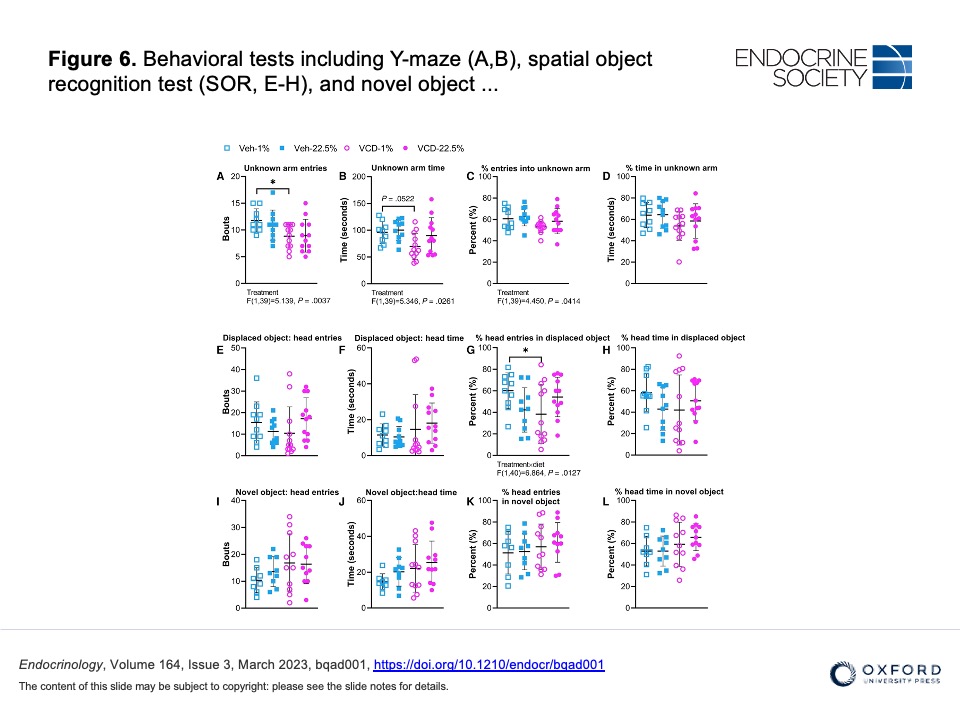
Figure 6. Behavioral tests including Y-maze (A,B), spatial object recognition test (SOR, E-H), and novel object recognition (NOR, I-L). (A) Unknown arm entries. (B) Unknown arm time. (C) Percent entries into the unknown arm. (D) Percent time in the unknown arm. (E) Head entries in the area of displaced object. (F) Head time in the area of displaced object. (G) Percent of head entries in the area of displaced object. (H) Percent of head time in the area of displaced object. (I) Head entries in the area of novel object. (J) Head time in the area of novel object. (K) Percent of head entries in the area of novel object. (L) Percent of head time in the area of displaced object. Veh-treated groups, n = 10 and VCD-treated gr oups, n = 12. Differences were determined by 2-way ANOVA followed with Holm–Sidak post hoc comparison (*P < .05) between and within treatment groups. Significant ANOVA results are shown under graph.
Unless provided in the caption above, the following copyright applies to the content of this slide: © The Author(s) 2023. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For permissions, please e-mail: journals.permissions@oup.comThis article is published and distributed under the terms of the Oxford University Press, Standard Journals Publication Model (https://academic.oup.com/journals/pages/open_access/funder_policies/chorus/standard_publication_model)
Ke Sui and others, Coconut Oil Saturated Fatty Acids Improved Energy Homeostasis but not Blood Pressure or Cognition in VCD-Treated Female Mice, Endocrinology, Volume 164, Issue 3, March 2023, bqad001, https://doi.org/10.1210/endocr/bqad001
Abstract:
Obesity, cardiometabolic disease, cognitive decline, and osteoporosis are symptoms of postmenopause, which can be modeled using 4-vinylcyclohexene diepoxide (VCD)–treated mice to induce ovarian failure and estrogen deficiency combined with high-fat diet (HFD) feeding. The trend of replacing saturated fatty acids (SFAs), for example coconut oil, with seed oils that are high in polyunsaturated fatty acids, specifically linoleic acid (LA), may induce inflammation and gut dysbiosis, and worsen symptoms of estrogen deficiency. To investigate this hypothesis, vehicle (Veh)- or VCD-treated C57BL/6J mice were fed a HFD (45% kcal fat) with a high LA:SFA ratio (22.5%: 8%), referred to as the 22.5% LA diet, or a HFD with a low LA:SFA ratio (1%: 31%), referred to as 1% LA diet, for a period of 23 to 25 weeks. Compared with VCD-treated mice fed the 22.5% LA diet, VCD-treated mice fed the 1% LA diet showed lower weight gain and improved glucose tolerance. However, VCD-treated mice fed the 1% LA diet had higher blood pressure and showed evidence of spatial cognitive impairment. Mice fed the 1% LA or 22.5% LA diets showed gut microbial taxa changes that have been associated with a mix of both beneficial and unfavorable cognitive and metabolic phenotypes. Overall, these data suggest that consuming different types of dietary fat from a variety of sources, without overemphasis on any particular type, is the optimal approach for promoting metabolic health regardless of estrogen status.
March 2023
(Michael Manhart)
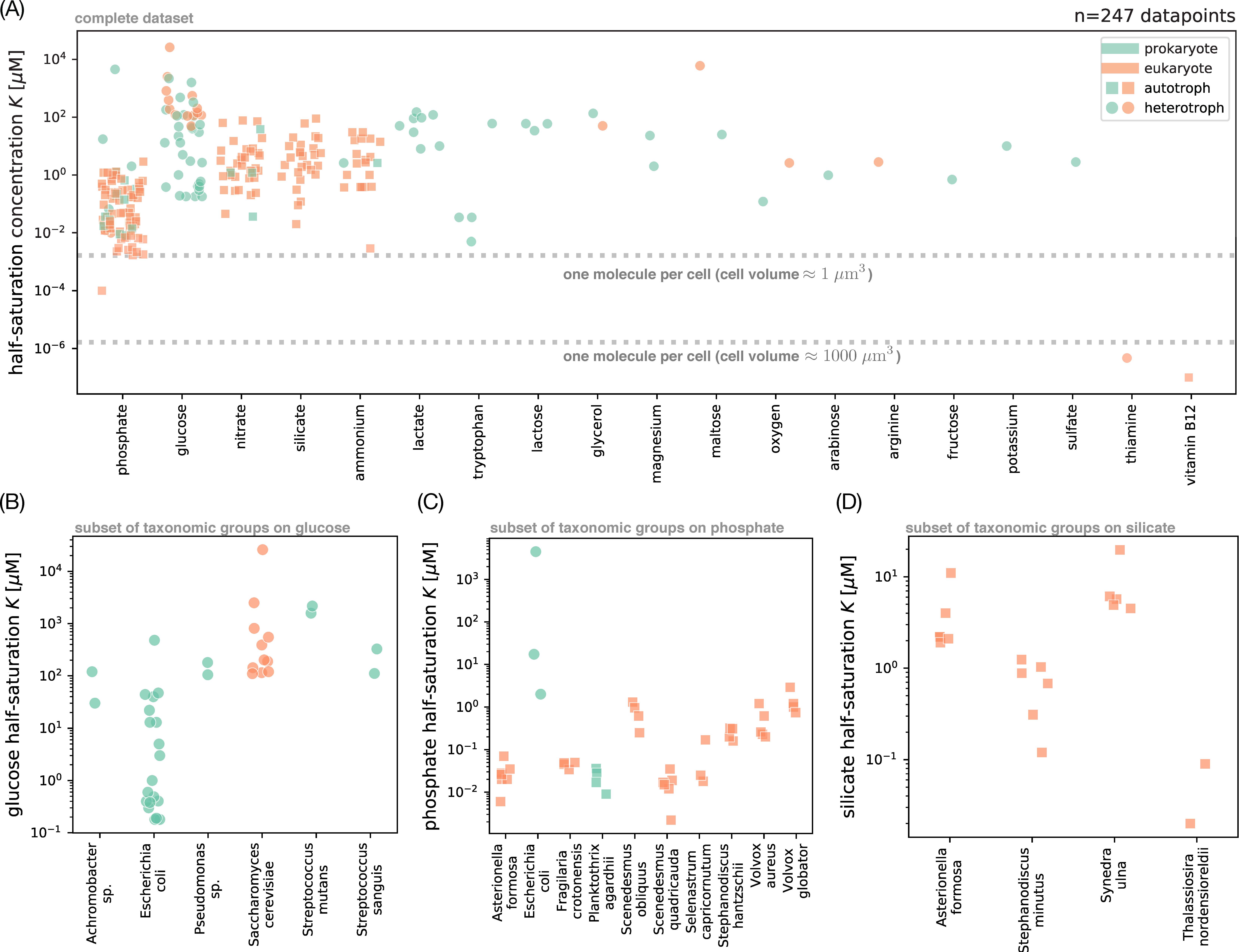
Fig. 2. Survey of measured half-saturation concentrations. (A) Complete set of half-saturation concentrations K for the Monod model of growth rate (Eq. 1) in our survey, grouped by resource (in decreasing order of number of data points). Each point represents a di erent measurement; color indicates whether the organism is a prokaryote (green) or eukaryote (orange), and shape indicates whether the organism can grow as an autotroph (square) or only as a heterotroph (circle). Dashed lines mark concentrations of one molecule per cell for approximate prokaryotic and eukaryotic cell volumes (45). (B) Subset of K measurements from panel A for glucose, grouped by taxon (only those with at least two measurements). We use the taxonomic identity given in the original publications, where an ending in sp. means that the isolate is a representative of the genus but was not identified at the species level. Symbols are the same as in panel A. For brevity, we use “glucose half-saturation” to refer to the half-saturation concentration for glucose as the limiting nutrient. (C) Subset of K measurements from panel A for phosphate, grouped by taxon (with at least three measurements). (D) Subset for silicate, grouped by taxon (with at least two measurements). SI Appendix for additional plots with K measurements for nitrate (SI Appendix, Fig. S4A) and ammonium (SI Appendix, Fig. S4B).
Fink JW, Held NA, Manhart M. Microbial population dynamics decouple growth response from environmental nutrient concentration. Proc Natl Acad Sci U S A. 2023 Jan 10;120(2):e2207295120. doi: 10.1073/pnas.2207295120. Epub 2023 Jan 4. PMID: 36598949; PMCID: PMC9926246. https://www.pnas.org/doi/10.1073/pnas.2207295120
Abstract:
How the growth rate of a microbial population responds to the environmental availability of chemical nutrients and other resources is a fundamental question in microbiology. Models of this response, such as the widely used Monod model, are generally characterized by a maximum growth rate and a half-saturation concentration of the resource. What values should we expect for these half-saturation concentrations, and how should they depend on the environmental concentration of the resource? We survey growth response data across a wide range of organisms and resources. We find that the half-saturation concentrations vary across orders of magnitude, even for the same organism and resource. To explain this variation, we develop an evolutionary model to show that demographic fluctuations (genetic drift) can constrain the adaptation of half-saturation concentrations. We find that this effect fundamentally differs depending on the type of population dynamics: Populations undergoing periodic bottlenecks of fixed size will adapt their half-saturation concentrations in proportion to the environmental resource concentrations, but populations undergoing periodic dilutions of fixed size will evolve half-saturation concentrations that are largely decoupled from the environmental concentrations. Our model not only provides testable predictions for laboratory evolution experiments, but it also reveals how an evolved half-saturation concentration may not reflect the organism's environment. In particular, this explains how organisms in resource-rich environments can still evolve fast growth at low resource concentrations. Altogether, our results demonstrate the critical role of population dynamics in shaping fundamental ecological traits.
Keywords: Monod model; half-saturation concentration; microbial evolution; resource competition; selection–drift balance.
February 2023
(Kenneth Yan
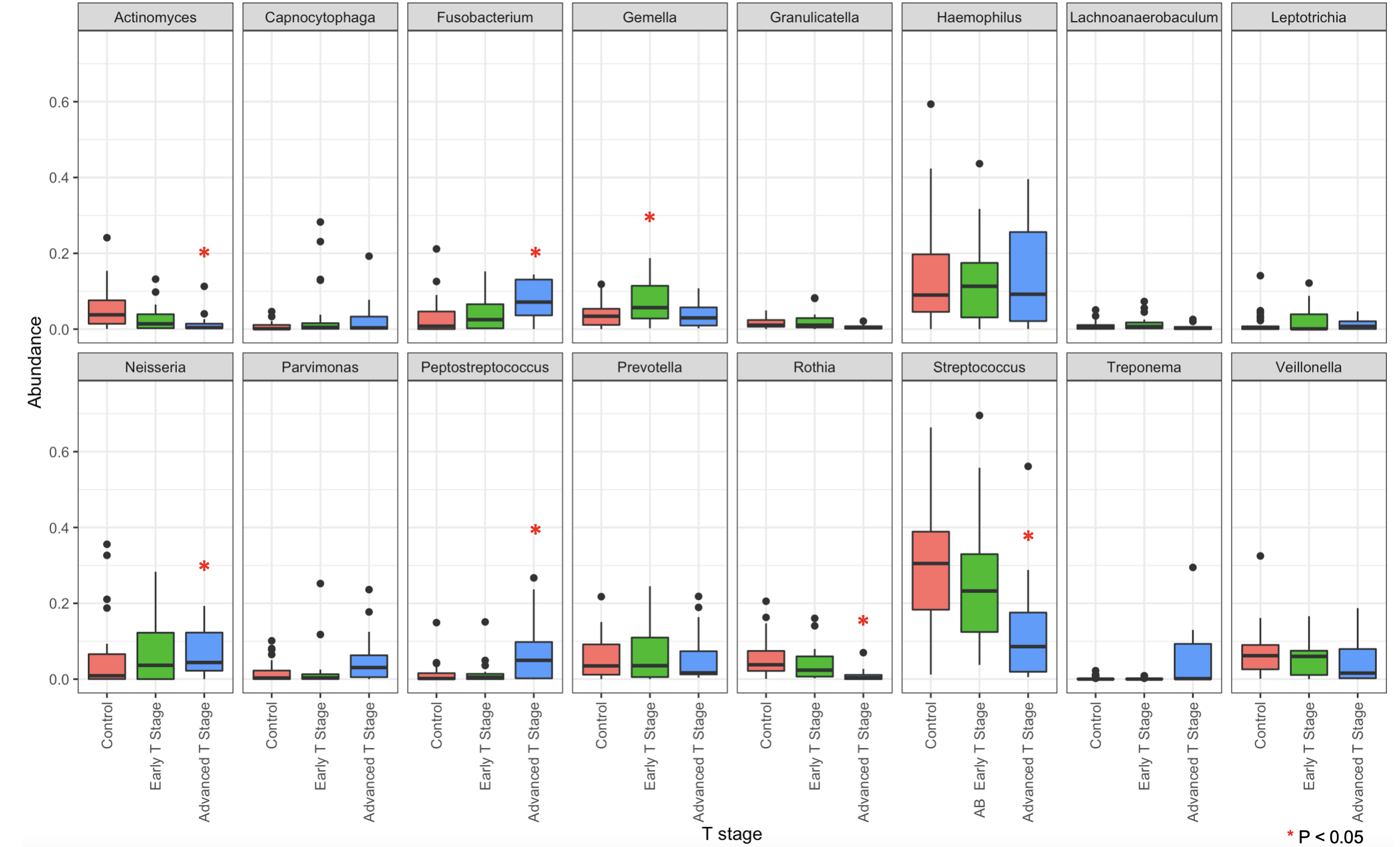
Figure 4c. Distribution of bacterial genera. Box plots for individual genera segregated by controls, early-stage (T1/T2) oral cancer and late-stage (T3/T4) cancer (*p<0.05).
Yan, K., Auger, S., Diaz, A., Naman, J., Vemulapalli, R., Hasina, R., Izumchenko, E., Shogan, B. and Agrawal, N. (2023), Microbial Changes Associated With Oral Cavity Cancer Progression. Otolaryngol Head Neck Surg. https://doi.org/10.1002/ohn.211
Abstract:
Objective: To examine the oral microbiome in the context of oral cavity squamous cell carcinoma.
Study Design: Basic science research.
Setting: Academic medical center.
Methods: Oral swabs were collected from patients presenting to the operating room for management of oral cavity squamous cell carcinoma and from age- and sex-matched control patients receiving surgery for unrelated benign conditions. 16S ribosomal RNA (rRNA) sequencing was performed on genetic material obtained from swabs. A bacterial rRNA gene library was created and sequence reads were sorted into taxonomic units.
Results: Thirty-one control patients (17 males) and 35 cancer patients (21 males) were enrolled. Ages ranged from 23 to 89 (median 63) for control patients and 35 to 86 (median 66) for cancer patients. Sixty-one percent of control patients and 63% of cancer patients were smokers. 16S analyses demonstrated a significant decrease in Streptococcus genera in oral cancer patients (34.11% vs 21.74% of the population, p = .04). Increases in Fusobacterium, Peptostreptococcus, Parvimonas, and Neisseria were also found. The abundance of these bacteria correlated with tumor T-stage.
Conclusion: 16S rRNA sequencing demonstrated changes in bacterial populations in oral cavity cancer and its progression compared to noncancer controls. We found increases in bacteria genera that correspond with tumor stage—Fusobacteria, Peptostreptococcus, Parvimonas, Neisseria, and Treponema. These data suggest that oral cancer creates an environment to facilitate foreign bacterial growth, rather than implicating a specific bacterial species in carcinogenesis. These bacteria can be employed as a potential marker for tumor progression or interrogated to better characterize the tumor microenvironment.
February 2023
(Nicole Fahrenfeld)
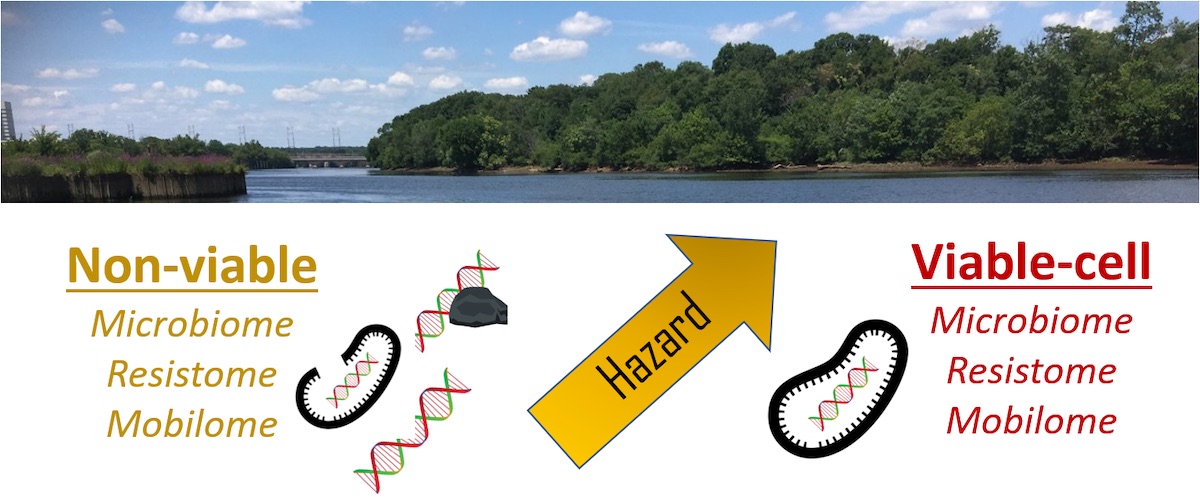
A.S. Deshpande, N.L. Fahrenfeld, Influence of DNA from non-viable sources on the riverine water and biofilm microbiome, resistome, mobilome, and resistance gene host assignments, Journal of Hazardous Materials, Volume 446,
2023, 130743, ISSN 0304-3894,
https://doi.org/10.1016/j.jhazmat.2023.130743.
(https://www.sciencedirect.com/science/article/pii/S0304389423000250)
Abstract: Shotgun metagenomic studies have revealed the diversity, relative abundance, and hosts of antibiotic resistance genes (ARGs) across environmental matrices. There is motivation to combine this method with viability-based techniques to better define ARG hazard. The objectives of this study were to evaluate the performance of different methods for extracellular DNA (eDNA) and putative “non-viable” cell DNA separation to understand the influence on ARG-host assignments. Paired water and biofilm samples were collected along a land use gradient. To study putative “viable-cell” DNA, samples were treated with propidium monoazide (i.e., PMA-DNA). To study eDNA, intracellular and extracellular DNA were separated. qPCR revealed differences in total 16S rRNA gene copies in water for filter vs. centrifuge-concentrated samples, but otherwise there were no differences in gene copy concentrations between DNA fractions. Next, metagenomic sequencing was performed on PMA-DNA and total DNA extracts revealing significant differences between the two for bacterial community structure and ARG profiles. Putative viable taxa containing pathogenic ARG hosts were identified in biofilm and water. Removing PMA-bound DNA improved N50 and assembly mapping compared to total DNA extracts. This study demonstrates the impact of different sample preparation methods on informing the hazard potential associated with riverine ARGs in water and biofilm.
Keywords: Propidium monoazide; ARG; Metagenomics; eDNA
January 2023
(Nicole Fahrenfeld)
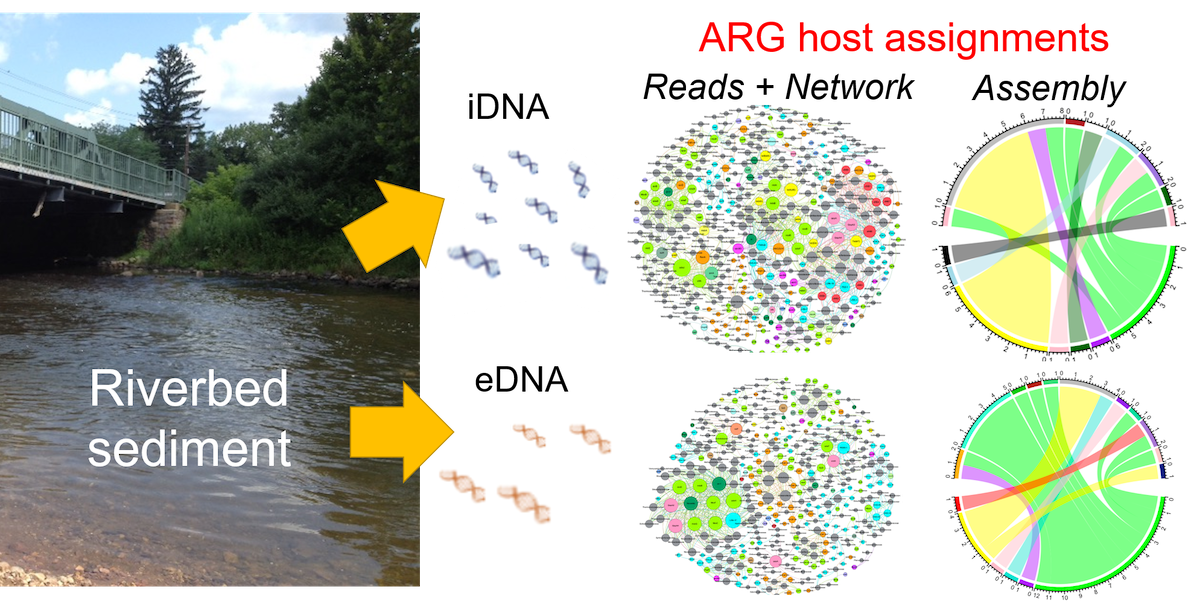
A.S. Deshpande, N.L. Fahrenfeld. Abundance, diversity, and host assignment of total, intracellular, and extracellular antibiotic resistance genes in riverbed sediments. Water Research. Volume 217, 2022. 118363, ISSN 0043-1354,
https://doi.org/10.1016/j.watres.2022.118363.
(https://www.sciencedirect.com/science/article/pii/S0043135422003256)
Abstract:
Human health risk assessment for environmental antibiotic resistant microbes requires not only quantifying the abundance of antibiotic resistance genes (ARGs) in environmental matrices, but also understanding their hosts and genetic context. Further, differentiating ARGs in intracellular and extracellular DNA (iDNA and eDNA) fractions may help refine our understanding of ARG transferability. The objectives of this study were to understand the (O1) abundance and diversity of extracellular, intracellular, and total ARGs along a land use gradient and (O2) impact of bioinformatics pipeline on the assignment of putative hosts for the ARGs observed in the different DNA fractions. Sediment samples were collected along a land use gradient in the Raritan River, New Jersey, USA. DNA was extracted to separate eDNA and iDNA and qPCR was performed for select ARGs and the 16S rRNA gene. Shotgun metagenomic sequencing was performed on DNA extracts for the different DNA fractions. ARG hosts were assigned via two different bioinformatic pipelines: network analysis of raw reads versus assembly. Results of the two pipelines were compared to evaluate their performance in terms of number and diversity of linkages and accuracy of in silico matrix spike host assignments. No differences were observed in the 16S rRNA gene normalized sul1 concentrations between the DNA fractions. The overall microbial community structure was more similar for iDNA and total DNA compared to eDNA and generally clustered by sampling site. ARGs associated with mobile genetic elements increased in iDNA for the downstream sites. Regarding host assignment, the raw reads pipeline via network analysis identified 247 ARG hosts as compared to 53 hosts identified by assembly pipeline. Other comparisons between the pipelines were made including ARG assignment to taxa containing waterborne pathogens and practical considerations regarding processing time.
Keywords: ARG; sul1; Metagenomic sequencing; Assembly; Network analysis; eDNA; iDNA
January 2023
(Sara Campbell)

Longoria, C. R., Guers, J. J., & Campbell, S. C. (2022). The Interplay between Cardiovascular Disease, Exercise, and the Gut Microbiome. Reviews in Cardiovascular Medicine, 23(11), 365.
Abstract:
Cardiovascular disease (CVD) is the leading cause of death worldwide, with physical inactivity being a known contributor to the global rates of CVD incidence. The gut microbiota has been associated with many diseases including CVD and other comorbidities such at type 2 diabetes and obesity. Researchers have begun to examine the gut microbiome as a predictor of early disease states by detecting disruptions, or dysbiosis, in the microbiota. Evidence is lacking to investigate the potential link between the gut microbiota, exercise, and CVD risk and development. Research supports that diets with whole food have reduced instances of CVD and associated diseases, increased abundances of beneficial gut bacteria, and altered gut-derived metabolite production. Further, exercise and lifestyle changes to increase physical activity demonstrate improved health outcomes related to CVD risk and comorbidities and gut microbial diversity. It is difficult to study an outcome such as CVD when including multiple factors; however, it is evident that exercise, lifestyle, and the gut microbiota contribute to improved health in their own ways. This review will highlight current research findings and what potential treatments of CVD may be generated by manipulation of the gut microbiota and/or exercise.
Keywords: gut microbiota; inflammation; endothelial function; prebiotics; probiotics; metabolites; trimethylamine N-oxide (TMAO)
December 2022
(Lee Kerkhof)
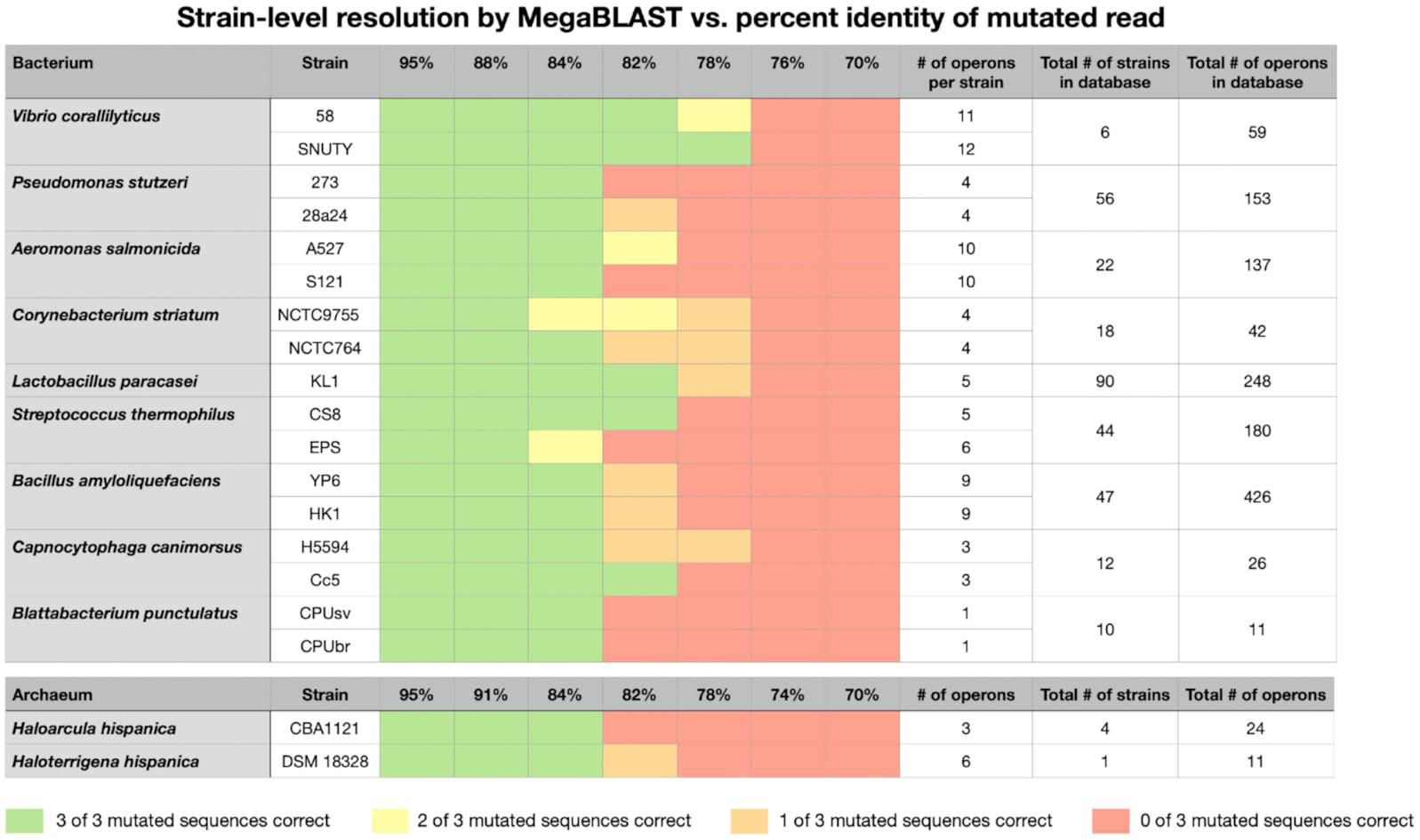
Figure 3. Heat map showing strain-level BLAST results from 19 in silico mutated sequences (in triplicate) vs % identity. The number of rRNA operons in each strain, the number of strains for each bacterial species and the total number of rRNA operons for each bacterial species in the database are indicated.
Lee J Kerkhof, Pierce A Roth, Samir V Deshpande, R Cory Bernhards, Alvin T Liem, Jessica M Hill, Max M Häggblom, Nicole S Webster, Olufunmilola Ibironke, Seda Mirzoyan, James J Polashock, Raymond F Sullivan, A ribosomal operon database and MegaBLAST settings for strain-level resolution of microbiomes, FEMS Microbes, Volume 3, 2022, xtac002, https://doi.org/10.1093/femsmc/xtac002
Abstract:
Current methods to characterize microbial communities generally employ sequencing of the 16S rRNA gene (<500 bp) with high accuracy (∼99%) but limited phylogenetic resolution. However, long-read sequencing now allows for the profiling of near-full-length ribosomal operons (16S-ITS-23S rRNA genes) on platforms such as the Oxford Nanopore MinION. Here, we describe an rRNA operon database with >300 ,000 entries, representing >10 ,000 prokaryotic species and ∼ 150, 000 strains. Additionally, BLAST parameters were identified for strain-level resolution using in silico mutated, mock rRNA operon sequences (70–95% identity) from four bacterial phyla and two members of the Euryarchaeota, mimicking MinION reads. MegaBLAST settings were determined that required <3 s per read on a Mac Mini with strain-level resolution for sequences with >84% identity. These settings were tested on rRNA operon libraries from the human respiratory tract, farm/forest soils and marine sponges ( n = 1, 322, 818 reads for all sample sets). Most rRNA operon reads in this data set yielded best BLAST hits (95 ± 8%). However, only 38–82% of library reads were compatible with strain-level resolution, reflecting the dominance of human/biomedical-associated prokaryotic entries in the database. Since the MinION and the Mac Mini are both portable, this study demonstrates the possibility of rapid strain-level microbiome analysis in the field.
November 2022
(Sandeep Yadav)
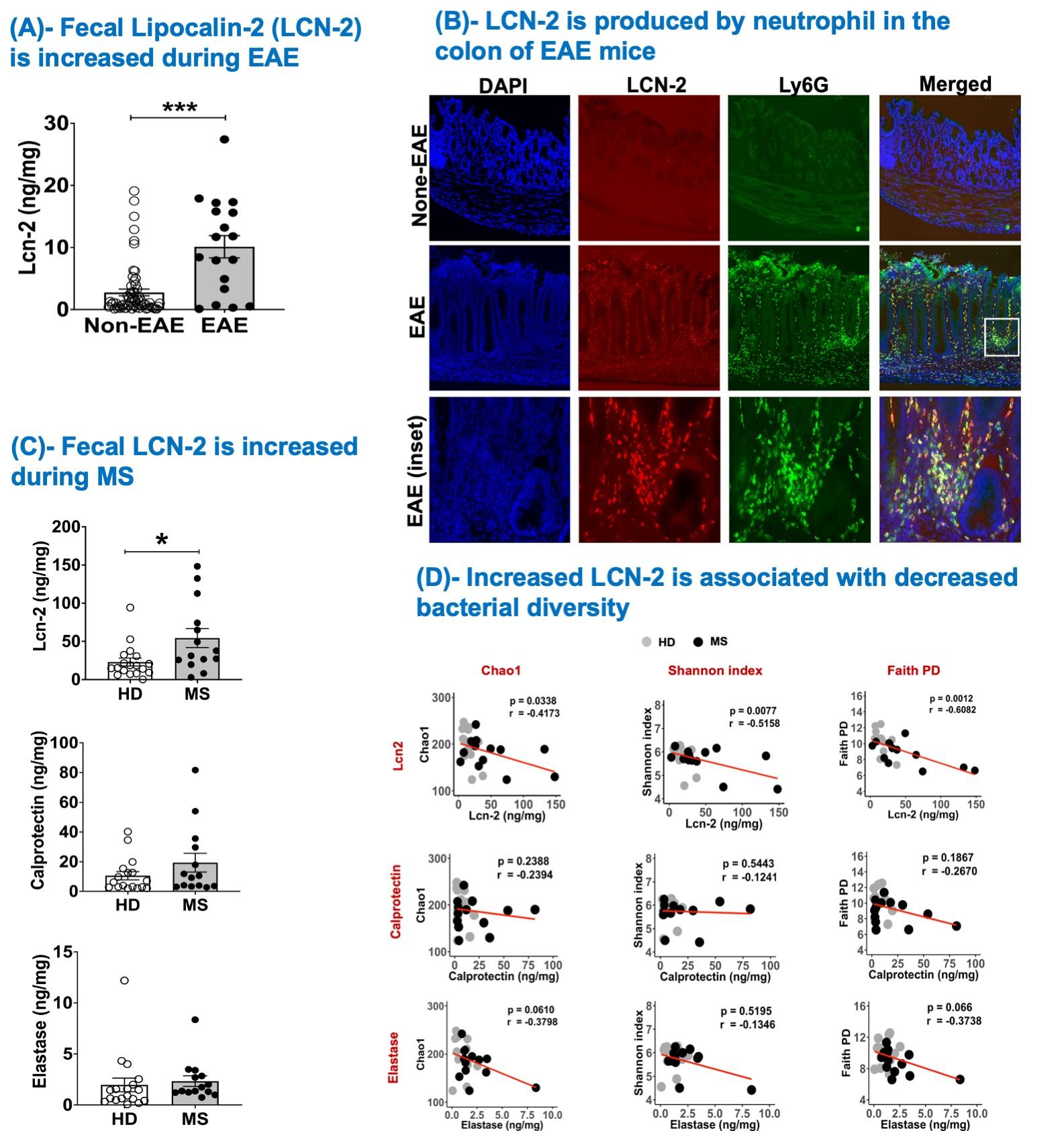
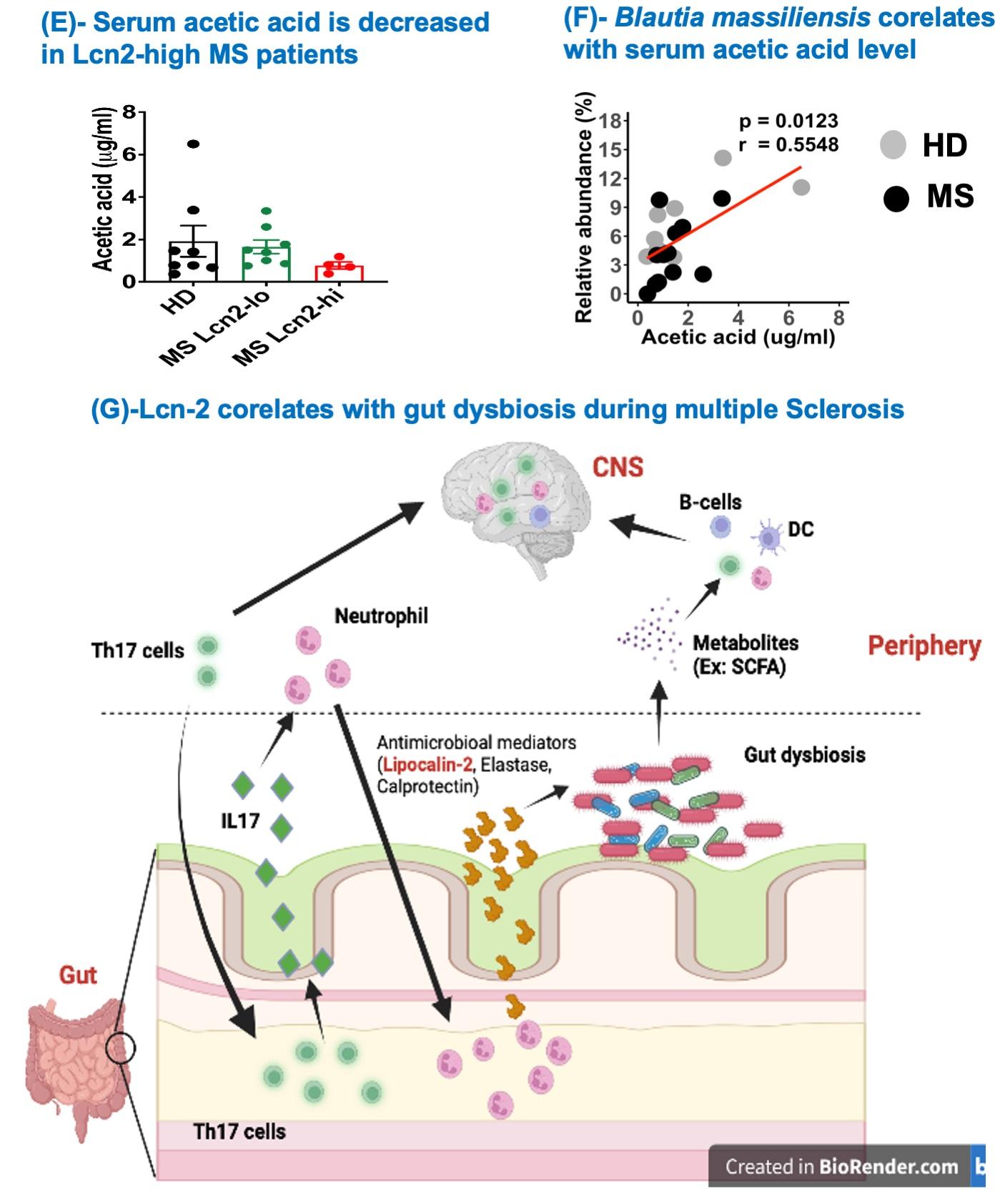
Figure 2. Fecal Lcn-2 level is a sensitive biological indicator for gut dysbiosis and intestinal inflammation in multiple sclerosis
Yadav SK, Ito N, Mindur JE, Kumar H, Youssef M, Suresh S, Kulkarni R, Rosario Y, Balashov KE, Dhib-Jalbut S and Ito K (2022) Fecal Lcn-2 level is a sensitive biological indicator for gut dysbiosis and intestinal inflammation in multiple sclerosis. Front. Immunol. 13:1015372.
https://doi.org/10.3389/fimmu.2022.1015372
(https://www.frontiersin.org/articles/10.3389/fimmu.2022.1015372/full)
Abstract: Multiple Sclerosis (MS) has been reported to be associated with intestinal inflammation and gut dysbiosis. To elucidate the underlying biology of MS-linked gut inflammation, we investigated gut infiltration of immune cells during the development of spontaneous experimental autoimmune encephalomyelitis (EAE) in humanized transgenic (Tg) mice expressing HLA-DR2a and human T cell receptor (TCR) specific for myelin basic protein peptide (MBP87-99)/HLA-DR2a complexes. Strikingly, we noted the simultaneous development of EAE and colitis, suggesting a link between autoimmune diseases of the central nervous system (CNS) and intestinal inflammation. Examination of the colon in these mice revealed the infiltration of MBP-specific Th17 cells as well as recruitment of neutrophils. Furthermore, we observed that fecal Lipocalin-2 (Lcn-2), a biomarker of intestinal inflammation, was significantly elevated and predominantly produced by the gut-infiltrating neutrophils. We then extended our findings to MS patients and demonstrate that their fecal Lcn-2 levels are significantly elevated compared to healthy donors (HDs). The elevation of fecal Lcn-2 levels correlated with reduced bacterial diversity and increased levels of other intestinal inflammation markers including neutrophil elastase and calprotectin. Of interest, bacteria thought to be beneficial for inflammatory bowel disease (IBD) such as Anaerobutyricum, Blautia, and Roseburia, were reduced in fecal Lcn-2-high MS patients. We also observed a decreasing trend in serum acetate (a short-chain fatty acid) levels in MS Lcn-2-high patients compared to HDs. Furthermore, a decrease in the relative abundance of Blautia massiliensis was significantly associated with a reduction of acetate in the serum of MS patients. This study suggests that gut infiltration of Th17 cells and recruitment of neutrophils are associated with the development of gut dysbiosis and intestinal inflammation, and that fecal Lcn-2 level is a sensitive biological indicator for gut dysbiosis in multiple sclerosis.
October 2022
(Luo Jia)
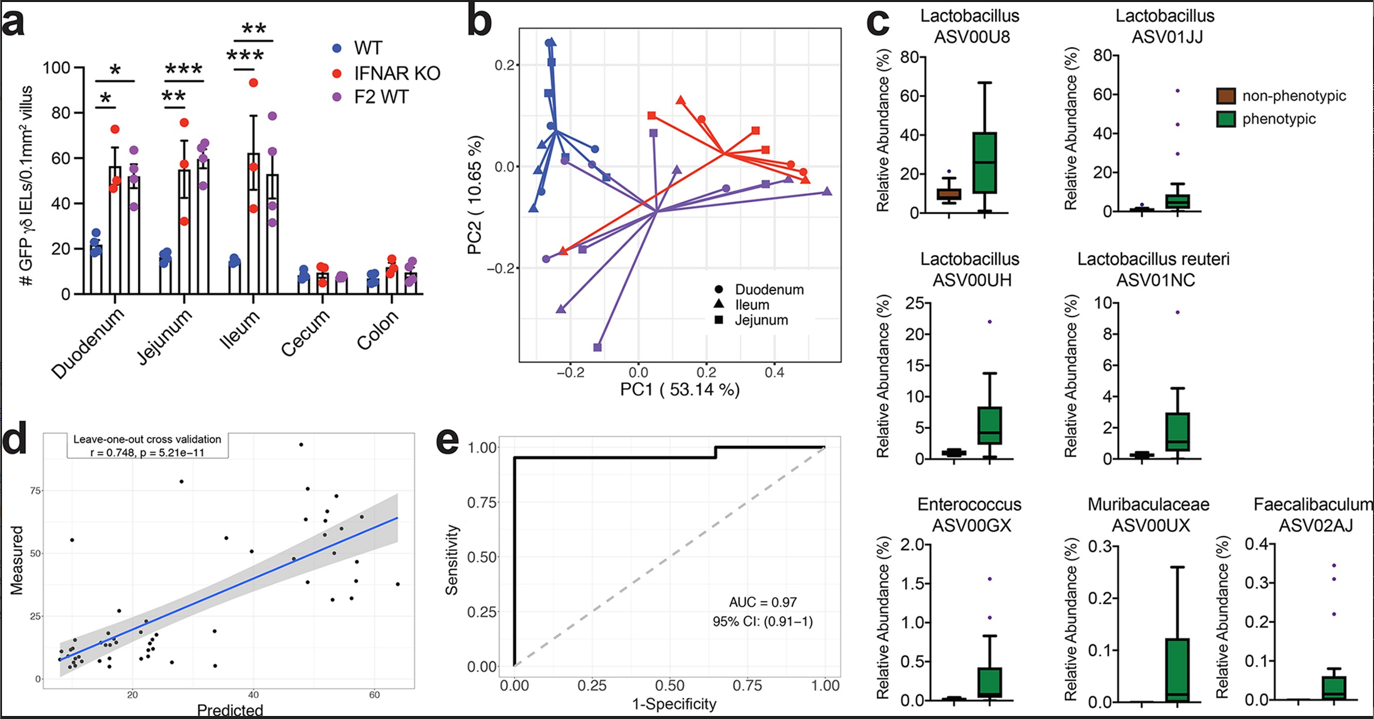
Figure 4. Small intestinal microbiota signature is associated with the local expansion of the γδ IEL compartment.
a Morphometric analysis of the number of GFP+ γδ T cells in untreated WT-S, IFNAR KO-S or F2 WT mice. b Principal coordinates analysis of 16S rRNA sequencing of SI luminal microbiota from WT-S, IFNAR KO-S and F2 WT mice. Bray Curtis distance was applied. c Seven ASVs in the SI were enriched in the mice with γδ IEL hyperproliferative phenotype. d and e Associations between the 7 ASVs in the SI enriched by the mice with γδ IEL hyperproliferative phenotype. Statistical analysis: a: one-way ANOVA with Tukey’s post hoc test; c: MaAsLin2 was applied to explore the differential ASVs in SI samples between mice with (IFNAR KO-S and F2 WT) and without the γδ IEL hyperproliferative phenotype (WT-S). BH-adjusted p values < 0.05 considered as significant. Boxes show the medians and the interquartile ranges (IQRs), the whiskers denote the lowest and highest values that were within 1.5 times the IQR from the first and third quartiles, and outliers are shown as individual points; d: Random Forest model with leave-one-out cross-validation was applied to use the ASVs abundance in the intestine to regress the intestinal γδ IEL cell number or e: to classify intestinal segments based on the γδ IEL hyperproliferative phenotype. Pearson correlation was used to compare the predicted and measured values. *P<0.05, **P<0.01, *** P<0.001.
Jia L, Wu G, Alonso S, Zhao C, Lemenze A, Lam YY, Zhao L, Edelblum KL. A transmissible γδ intraepithelial lymphocyte hyperproliferative phenotype is associated with the intestinal microbiota and confers protection against acute infection. Mucosal Immunol. 2022 Apr;15(4):772-782. doi: 10.1038/s41385-022-00522-x. Epub 2022 May 19. PMID: 35589986; PMCID: PMC9262869.
doi: 10.1038/s41385-022-00522-x
(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9262869/)
Abstract:
Intraepithelial lymphocytes expressing the gamma delta T cell receptor (gamma delta IELs) serve as a first line of defense against luminal microbes. Although the presence of an intact microbiota is dispensable for gamma delta IEL development, several microbial factors contribute to the maintenance of this sentinel population. However, whether specific commensals influence population of the gamma delta IEL compartment under homeostatic conditions has yet to be determined. We identified a novel gamma delta IEL hyperproliferative phenotype that is characterized by expansion of multiple Vgamma subsets. Horizontal transfer of this hyperproliferative phenotype to mice harboring a phenotypically normal gamma delta IEL compartment was prevented following antibiotic treatment, thus demonstrating that the microbiota is both necessary and sufficient for the observed increase in gamma delta IELs. Further, we identified two guilds of small intestinal or fecal bacteria represented by 12 amplicon sequence variants (ASV) that are strongly associated with gamma delta IEL expansion. Using intravital microscopy, we find that hyperproliferative gamma delta IELs also exhibit increased migratory behavior leading to enhanced protection against bacterial infection. These findings reveal that transfer of a specific group of commensals can regulate gamma delta IEL homeostasis and immune surveillance, which may provide a novel means to reinforce the epithelial barrier.
September 2022
(Bassel Ghaddar)
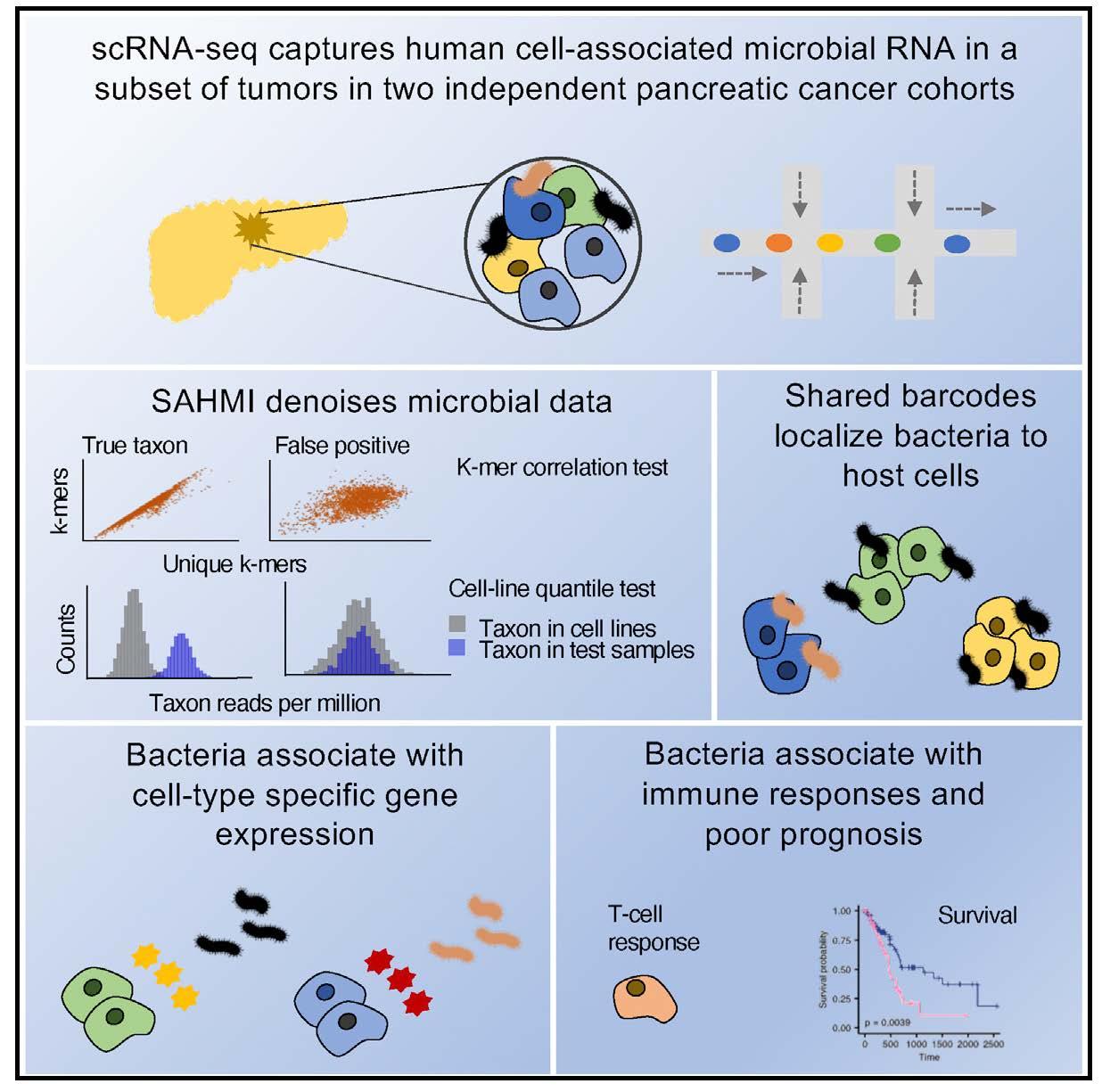
Graphical Abstract [see abstract below for more information]
Bassel Ghaddar, Antara Biswas, Chris Harris, M. Bishr Omary, Darren R. Carpizo, Martin J. Blaser, Subhajyoti De.
Tumor microbiome links cellular programs and immunity in pancreatic cancer, Cancer Cell, Volume 40, Issue 10, 2022, Pages 1240-1253.e5, ISSN 1535-6108, https://doi.org/10.1016/j.ccell.2022.09.009.
(https://www.sciencedirect.com/science/article/pii/S153561082200438X)
Abstract
Microorganisms are detected in multiple cancer types, including in putatively sterile organs, but the contexts in which they influence oncogenesis or anti-tumor responses in humans remain unclear. We recently developed single-cell analysis of host-microbiome interactions (SAHMI), a computational pipeline to recover and denoise microbial signals from single-cell sequencing of host tissues. Here we use SAHMI to interrogate tumor-microbiome interactions in two human pancreatic cancer cohorts. We identify somatic-cell-associated bacteria in a subset of tumors and their near absence in nonmalignant tissues. These bacteria predominantly pair with tumor cells, and their presence is associated with cell-type-specific gene expression and pathway activities, including cell motility and immune signaling. Modeling results indicate that tumor-infiltrating lymphocytes closely resemble T cells from infected tissue. Finally, using multiple independent datasets, a signature of cell-associated bacteria predicts clinical prognosis. Tumor-microbiome crosstalk may modulate tumorigenesis in pancreatic cancer with implications for clinical management.
Keywords: cancer genomics; microbiome; immunity; oncogenesis; cancer biology; gene expression; single cell sequencing